
What is GAGE?
GAGE is an evaluation of the very latest large-scale genome assembly algorithms. We have organized this "bake-off" as an attempt to produce a realistic assessment of genome assembly software in a rapidly changing field of next-generation sequencing. The main results of GAGE have now been published in the journal Genome Research: GAGE: A critical evaluation of genome assemblies and assembly algorithms.
Why do we need GAGE?
The rapidly falling cost of sequencing means that many scientists can now contemplate whole-genome sequencing of their favorite organism. Interest in genome sequencing of new species has increased rapidly, inspired by a few high-profile successes such as the panda genome. However, assembling large genomes from short reads remains a very challenging problem, and except for a limited number of specialists in genome assembly, very few scientists know how to design and execute such a project. The GAGE competition will provide a snapshot, using the latest short-read technology, of how the current genome assemblers compare on a sample of large-scale sequencing projects. This will help answer questions such as:
- How much sequencing coverage do I need for my genome project?
- What can I expect the resulting assembly to look like?
- (Most important) Which assembly software should I use?
- What parameters should I use when I run the software?
How does GAGE differ from the other assembly bake-offs?
At least two other assembly comparisons have been announced for 2011, the Assemblathon (http://assemblathon.org/) and dnGASP (http://cnag.bsc.es/). Here are some differences:
- GAGE is being run by assembly experts. Our team has assembled hundreds of genomes, and has written some of the leading genome assembly software. We have been evaluating assemblers for more than 10 years. All of the assemblies and the comparisons among them will be conducted by experts.
- All natural ingredients. GAGE will use FOUR different whole-genome shotgun data sets, all from recent sequencing projects. Assemblathon and dnGASP will both use simulated data. Who can say how simulated data relates to real results? We prefer the real thing.
- Completely open protocols. We will compare multiple genome assembly programs, and we will describe all the parameters used to run them. We will also describe all the steps we take in cleaning up the data (pre-processing) and scrubbing the output of the assembly programs (post-processing). All of our results will thus be reproducible by anyone with sufficient computing resources.
What genomes will you use?
We have identified four whole-genome shotgun sequence data sets for this competition, representing a wide phylogenetic range and differing degrees of difficulty. All data sets will be Illumina reads only, although we have additional data from 454 and Sanger sequencing to use as validation. The genomes are:
These genomes range in size from just 2 Mbp (S. aureus) to 3 Gbp (human).
- Staphylococcus aureus
- Rhodobacter sphaeroides
- Human (e.g. chromosome 14)
- Bombus impatiens (a species of bee)
Who is organizing GAGE?
The GAGE competition has been organized by several groups, all of them specialists in genome assembly:
- Steven Salzberg (JHU)
- Adam Phillippy (National Biodefense Analysis and Countermeasures Center)
- Mihai Pop (CBCB,UMD)
- Michael Schatz (Cold Spring Harbor Laboratory)
- Jim Yorke (IPST,UMD)
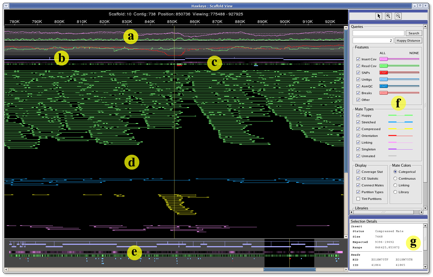
The scaffold view from the Hawkeye genome assembly viewer displays (a) statistical information, (b) scaffolded contigs, (c) feature tracks, (d) inserts, (e) overview panel, (f) control panel, and (g) details panel. Inserts are colored by category (green→happy, blue→stretched, yellow→compressed).
The evaluation and assembly of all genomes will be conducted by members of the organizing committee and their research groups.
Acknowledgments
We are grateful to our collaborators and to other genome sequencing
groups for making sequence data freely available. Special thanks
to Gene Robinson (University of Illinois) for sharing the Bombus impatiens genome data, which
was supported by a 2009 NIH Director's Pioneer Award (1DP1OD006416) to
Prof. Robinson. We are equally
grateful to the developers of the assembly software systems used in
our evaluation for making their software freely available. (More
details to appear here later.) This work is supported in part by NIH
grant R01-LM006845 (S. Salzberg), R01-HG04885 (M. Pop), R01-HG002945
(J. Yorke), and by USDA NRI grant 2009-35205-05209 (National Institute
of Food and Agriculture) to Salzberg and Yorke.